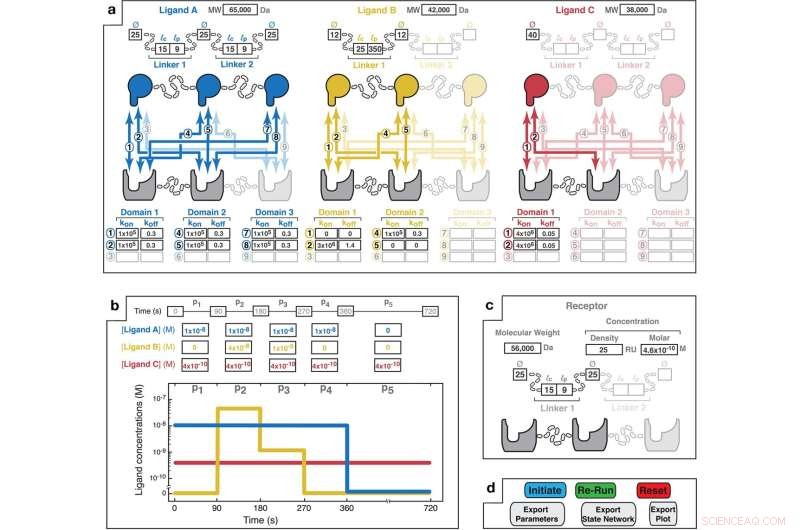
MVsim-ingångsdesigngränssnittet tillhandahåller interaktiv parameterspecifikation för system med multivalent, multimolekylär interaktion. a Ett peka-och-klicka-gränssnitt gör det möjligt för användaren att välja antalet ligander (upp till tre) och valenser för liganden(er) och receptorn (upp till trivalent) som utgör det multivalenta systemet. Baserat på den valda designen specificerar användaren strukturen för var och en av liganderna genom att ange tillämplig molekylvikt (MW); de bindande domänens diametrar (Ø); konturlängderna (lc för länkarna (d.v.s. det maximala avståndet från ände till ände; t.ex. 3,5 Å och 1,5 Å per aminosyra för en slumpmässig spole respektive alfahelix); och persistenslängderna (lp) för länkar. Vidare är de tillämpliga kombinatoriska interaktionerna (numrerade 1 till 9) unika för varje receptor-ligandparning framhävda. Parameterfält tillåter inmatning av monovalenta hastighetskonstanter för varje parvis interaktion. Icke-bindande interaktioner kan indikeras med k på och k av värden på noll (t.ex. som illustreras med ligand B i gult för interaktioner "1" och "5"). b Ett inmatningsfält låter användaren specificera mönster för de totala bulkligandkoncentrationerna. En associationsfas inträffar under perioder med icke-noll bulkligandkoncentration (t.ex. 90–270 s för Ligand B). Dissociationsfaser inträffar när liganden avlägsnas från bulklösningen (t.ex. 360–720 s för Ligand A). Här specificeras ligand C som kontinuerligt närvarande i lösning under 720 s av interaktionens tidsförlopp. Den grafiska displayen tillåter visualisering av det specificerade bulkkoncentrationspulsmönstret. c Användarinmatningsparametrar för receptorn. Receptorkoncentration kan specificeras som antingen en SPR-liknande ytdensitet (mätt i RU; där 1 RU är lika med ~1 pg/mm 2 ) eller en molär koncentration. Receptortopologi specificeras i samma form som beskrivits ovan för liganderna. d MVsim kontrollfliken möjliggör initiering, iteration och export av bindningssimuleringar. "Initiera" utför en simulering. "Re-run" exekverar en förkortad simulering som används när inga ändringar gjordes i valensen eller topologin för systemet. "Återställ" startar om appen och rensar användarinmatningsparametrar från alla fält. Kredit:Nature Communications (2022). DOI:10.1038/s41467-022-32496-6
Ett team under ledning av University of Minnesota Twin Cities biomedicinska ingenjörer har utvecklat en universellt tillgänglig applikation som kan simulera komplexa molekylära interaktioner, vilket gör det möjligt för forskare att utforma bättre behandlingar för sjukdomar som cancer och covid-19.
Uppsatsen bygger på en studie som forskarna publicerade 2019. Nu har de utökat tekniken för att simulera ännu mer komplexa molekylära interaktioner, gjort applikationen enkel att använda för icke-experter och tillämpat sina resultat för att belysa hur SARS -CoV-2-virus infekterar kroppen.
Studien publiceras i Nature Communications , och appen, som heter MVsim, är fritt tillgänglig för andra forskare på GitHub.
Simulatorn förutsäger styrkan, hastigheten och selektiviteten hos multivalenta interaktioner, som involverar molekyler som har flera bindningsställen och kan användas för att utveckla läkemedel mot sjukdomar, särskilt cancer och covid-19.
"Multivalenta interaktioner är verkligen viktiga i naturliga biologiska system, och de börjar nu på ett kreativt sätt utnyttjas för att skapa nya terapeutiska läkemedel som utnyttjar deras unika bindningsegenskaper", säger Casim Sarkar, senior författare till artikeln och professor vid University of Minnesota Institutionen för biomedicinsk teknik.
"Med multivalenta läkemedel kan man i princip rikta in sig på celler mycket specifikt på ett sätt som inte är möjligt med vanliga monovalenta läkemedel, men det finns många variabler att ta hänsyn till i deras design och mycket av arbetet på området hittills har gjorts. genom experimentellt försök och fel," tillade Sarkar. "Nu, med hjälp av MVsim, kan vi göra bra förutsägelser som kan användas för att mer rationellt utforma sådana terapier."
Många cancerläkemedel binder inte bara till tumörceller utan även till celler som de inte är avsedda att rikta in sig på, vilket ofta skapar oönskade biverkningar för patienten. Genom att optimera specificiteten för multivalenta interaktioner med MVsim kan forskare designa läkemedel som mer specifikt riktar sig mot cellerna i en tumör samtidigt som bindningen till andra celler i kroppen minimeras.
Ett annat exempel är SARS-CoV-2-viruset. Forskare vet att viruset utvecklas för att bättre infektera våra celler och undvika vårt immunsystem, men de molekylära mekanismerna bakom hur viruset gör detta är relativt okända. Med hjälp av sin MVsim-teknik kunde forskarna vid University of Minnesota utforska denna process mer på djupet och avslöja hastigheten med vilken individuella bindningsdomäner inom virusets multivalenta spikprotein växlar mellan ett cellinfekterande tillstånd och ett immunundvikande tillstånd.
"We essentially have a computational microscope that allows us to look under the hood and see what multivalent proteins such as the SARS-CoV-2 spike protein are doing at a molecular level," Sarkar explained. "This level of molecular detail is hard to capture with a physical experiment. One of the real powers of MVsim is that we can not only learn more about how these systems work but we can also use this tool to design new multivalent interactions for diseases like cancer and COVID-19."
The researchers have already identified potential ways to limit the infectivity of current and future SARS-CoV-2 variants, which they plan to test soon. + Utforska vidare