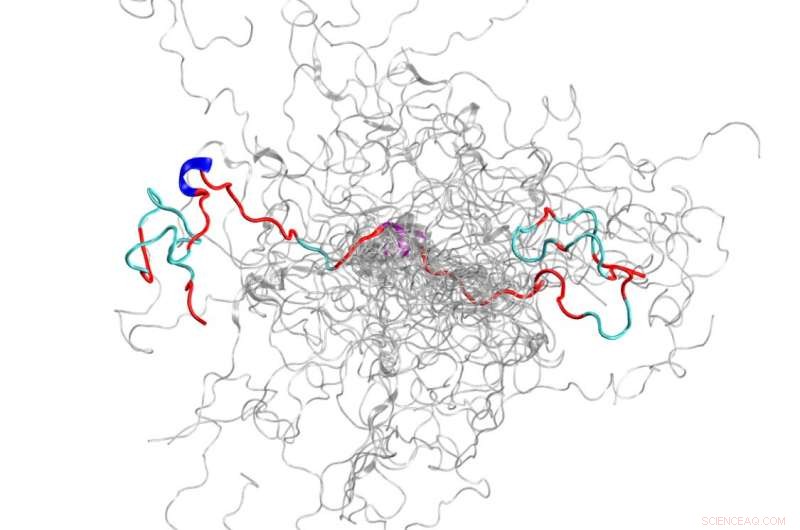
Konfigurationsensemblen (en samling 3D-strukturer) av ett i sig oordnat protein, N-terminalen av c-Src kinas, som är ett viktigt signalprotein hos människor. Kredit:Oak Ridge National Laboratory, USA:s energidepartement
Genom att använda superdatorn Titan och spallationsneutronkällan vid Department of Energy's Oak Ridge National Laboratory, forskare har skapat den mest exakta 3D-modellen hittills av ett i sig stört protein, avslöjar ensemblen av dess strukturer på atomnivå.
Som namnet antyder, en internflykting antar inte en beordrad, statisk struktur som andra proteiner; istället, den är flexibel och kan använda flera 3D-strukturer. Denna brist på en unik struktur är nödvändig för IDP:s biologiska funktion men gör det tekniskt utmanande att studera. IDP:er kan vara ett helt protein eller en domän av ett annars strukturerat protein, och de utgör en stor del av människor, mikrob, och växtproteiner.
Loukas Petridis, en stabsforskare vid Center for Molecular Biophysics vid ORNL, har riktat ett team av forskare till ett nytt sätt att skapa exakta fysiska modeller av sådana flexibla biosystem, vilket kan leda till en bättre förståelse av deras biologiska funktioner. Under de senaste tre åren, teamet har kombinerat neutronspridningsexperiment med förbättrad sampling molekylär dynamik (MD) simuleringar som är så beräkningskrävande att de krävde bearbetningskraften hos Titan, den nyligen avvecklade 27 petaflop Cray XK7 vid Oak Ridge Leadership Computing Facility, en DOE Office of Science User Facility på ORNL.
"Att studera dessa internflyktingar är ganska svårt, ur både experiment- och modelleringsperspektiv, sade Utsab Shrestha, huvudförfattaren till lagets tidning, nyligen publicerad i Proceedings of the National Academy of Sciences . "Vi tänkte inte bara på det från experiment eller simulering enbart, vi planerade på ett sätt att vi skulle synergisera båda dessa tillvägagångssätt – kombinera dem på ett sätt så att vi kunde få mer exakt information om internflyktingar. Specifikt, simuleringar hjälpte oss att skapa en exakt ensemble av IDP vid atomär upplösning, vilket är svårt att avgöra från enbart experiment."
Vanligtvis, forskare genomför experiment som småvinklar neutronspridning, liten vinkel röntgenspridning, eller kärnmagnetisk resonans för att undersöka flexibla biologiska system. Dock, dessa metoder ger inte en detaljerad bild på atomnivå av en IDP:s 3D-strukturer, känd som dess konfigurationsensemble. Vidare, de kan bara producera ensemble-genomsnittsdata, snarare än de specifika underliggande proteinstrukturkonfigurationerna. Forskare har också utfört datorsimuleringar av IDP och jämfört dem med sådana experiment, i hopp om att få samma resultat för att verifiera noggrannheten hos deras modeller.
"Men det slutar med att de inte håller med om experimenten, " sa Petridis. "Och på grund av diskrepansen mellan simuleringarna och experimenten, de måste väga om simuleringarna – de måste justera simuleringsresultaten för att få dem att matcha experimenten, vilket är frustrerande. Det var det senaste fram till vårt arbete."
Dator-MD-simuleringar utförda av Shrestha använde förbättrade provtagningsmetoder som lyckades matcha inte bara neutronspridningsexperiment – utförda av Viswanathan Gurumoorthy och hans kollegor på SNS, en DOE Office of Science User Facility vid ORNL – men också tidigare publicerade NMR-data. Dessa MD-simuleringar använder fysik för att bestämma hur proteiner rör sig. Nyckeln till lagets framgång var att köra många MD-simuleringar parallellt på Titan, låter simuleringarna kommunicera med varandra och utbyta information.
"Detta är mycket viktigt eftersom det tillåter simuleringen att ta prov på ett större konfigurationsutrymme, utforska fler av de tredimensionella strukturerna på ett mer effektivt sätt, " sa Petridis. "Det är därför som denna förbättrade samplings-MD kan ge resultat som den normala MD-simuleringen inte kan. Vi skulle behöva köra en normal MD-simulering i flera år för att få samma resultat."
IDP som teamet valde att studera är den N-terminala domänen av c-Src kinas, som är ett viktigt signalprotein hos människor. Mutationer i detta komplexa protein har korrelerats med cancer, vilket också gör det till ett viktigt läkemedelsmål. När du kartlade denna tidigare grumliga domän, forskarna kunde upptäcka ny information om dess 3D-strukturer som tidigare metoder inte hade visat. Till exempel, även om det till stor del är oordnat, detta protein bildar övergående ordnade strukturer, såsom spiraler.
"Kombinationen av neutronspridningsexperiment och simulering är mycket kraftfull, "Sa Petridis. "Validering av simuleringarna i jämförelse med neutronspridningsexperiment är avgörande för att ha förtroende för simuleringsresultaten. De validerade simuleringarna kan sedan ge detaljerad information som inte erhålls direkt genom experiment."
Den detaljerade datormodellen av IDP:s 3D-strukturensemble öppnar dörren till mer experimenterande. Till exempel, forskare kunde simulera effekten av fosforylering (tillägget av en fosfatgrupp till proteinet som kan reglera proteinets funktion) för att se vilka strukturella förändringar som sker i c-Src-kinas som kan påverka dess funktion. Mutationers roll skulle också kunna undersökas:Om en forskare ändrar en aminosyra i kedjan, hur påverkar detta strukturen eller ensemblen av strukturer?
"Det finns många obesvarade frågor för c-Src kinas i synnerhet som skulle kunna besvaras när det gäller interaktioner med andra partners - effekten av fosforylering, effekten av mutationer, sa Petridis.
Utöver de potentiella vetenskapliga användningsområdena för själva modellen, Petridis ser möjligheter att tillämpa användningen av högpresterande beräkningar för att köra förbättrad sampling MD för att studera strukturerna hos många andra viktiga IDP:er, som skulle kunna ge insikt om deras funktion. Och mer allmänt, teamet vill utveckla simuleringstekniker som kan reproducera små vinklar neutronspridningsprofiler för ännu mer komplexa biologiska system.
"Vi vill inte bara undersöka de oordnade proteinerna - vi vill ha mycket större system som innehåller ordnade och oordnade domäner som kan interagera med membran eller DNA, " sa Petridis. "Neutronspridning är, från min synvinkel, den bästa experimentella tekniken för att undersöka dessa flerkomponentsystem – till exempel, ett protein som interagerar med ett membran eller ett protein som interagerar med DNA. Men, fortfarande, neutronspridning behöver noggranna simuleringar för att bättre tolka data."