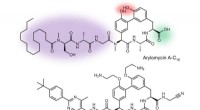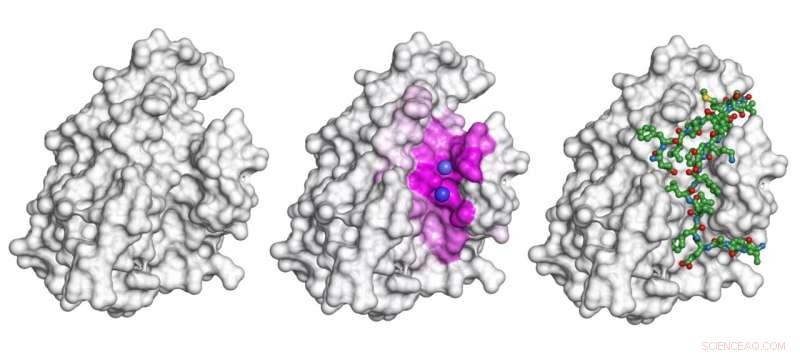
Den grå formen är ett protein. För scenariot med att detta protein binder till peptiden som visas som en grönaktig stick-and-ball-modell till höger, modellen som presenteras i studien framhäver ytan som är involverad i interaktionen (det rosa området i mitten) och förutsäger de exakta bindningsställena (lila sfärer). Kredit:Igor Kozlovskii och Petr Popov / Skoltech
Två Skoltech-forskare har presenterat en mycket effektiv neurala nätverksmodell som använder data om strukturen hos proteiner för att förutsäga vilka av deras delar som interagerar med andra biologiska molekyler som kallas peptider. Att veta att detta är användbart för att utveckla läkemedel baserade på peptider, som kan påverka protein-protein-interaktioner inom celler på ett riktat och ogiftigt sätt, reglerar ett brett spektrum av cellulära processer. Studien kom ut i Journal of Chemical Information and Modeling .
Proteiner är cellers maskineri, rör sig, umgås med varandra, och kör alla typer av operationer. Farmakologer har alltid varit fascinerade av möjligheten att mixtra med interaktionerna mellan proteiner. Ändå verkade de vara förbjudna som ett potentiellt läkemedelsmål:De större terapeutiska molekylerna, kallas biologiska läkemedel, kunde inte tränga in i cellen för att verka på proteiner, medan småmolekylära medel ofta visade sig vara oförmögna till sådan verkan.
Peptider, som naturligt förmedlar eller reglerar cirka 40 % av cellulära processer, ockupera en lovande medelväg och har utsikter för mediciner inriktade på protein-proteininteraktioner. Peptider erbjuder det bästa av två världar:Som små molekyler, de kan penetrera cellmembranet för att faktiskt nå sina mål, och de uppvisar också låg toxicitet, tillsammans med hög affinitet och specificitet (stark och fokuserad verkan) - kännetecknen för biologiska läkemedel.
För att designa peptidbaserade läkemedel, farmakologer behöver känna till de så kallade bindningsställena för ett visst proteinmål. Det är, fläckarna på proteinet som kan binda till en peptid. Ju fler sådana platser är kända, desto fler möjligheter finns för läkemedelsdesign.
Forskare kan identifiera bindningsställen experimentellt, till exempel, med hjälp av röntgenkristallografi, som avslöjar 3D-strukturen hos kristalliserade proteiner genom att studera hur de diffrakterar röntgenstrålar. Men detta är väldigt dyrt att göra för en lång lista med molekyler, och beräkningsmetoder erbjuder ett snabbare och billigare alternativ. Vissa av dem bygger på maskininlärningstekniker, och när mer data om strukturerna hos protein-peptidkomplex ackumuleras, dessa metoder blir mer kraftfulla och ger allt bättre förutsägelser om bindningsställen.
I deras 22 juli papper i Journal of Chemical Information and Modeling , Skoltech Ph.D. student Igor Kozlovskii och biträdande professor Petr Popov från iMolecule-gruppen presenterade en beräkningsmetod som heter BiteNetPp, som utnyttjar kraften i 3D-konvolutionella neurala nätverk för att detektera protein-peptidbindningsställen. I BiteNetPp, en känd proteinstruktur matas till ett neuralt nätverk, som sedan lyfter fram misstänkta peptidbindningsställen, och matar ut en uppsättning förmodade 3D-koordinater, tillsammans med tillhörande sannolikhetspoäng.
Petr Popov kommenterar tillvägagångssättet för att detektera bindningsställen som bildigenkänning, som ursprungligen introducerades i lagets tidigare artikel och överfördes till studien som rapporterades i denna berättelse:"Precis som neurala nätverk kan tränas att känna igen, säga, fotgängare eller cyklister i vanliga 2D-bilder, vi ser detektion av bindningsställen som att upptäcka en viss typ av objekt i en bild. Skillnaden är att vi använder 3D-atomstrukturdata som våra indata, så modellen fungerar på 'voxels, "en tredimensionell analog av pixlar."
Den nyligen presenterade modellen bygger faktiskt på den i föregående artikel. "Detta kallas domänanpassning. BiteNetPp är den första modellen som har finjusterats på en protein-peptiddatauppsättning efter att ha tränats på protein-småmolekyldata, " Popov förklarar. "Du kan föreställa dig detta som att träna en modell för att identifiera platser där cyklister tenderar att stanna på gatan, men du börjar med uppgifter om var fotgängare tenderar att stanna – och först därefter utökar du din domän till cyklister. Istället för att börja från början, du tränar om modellen, förutse att "bindningsplatserna" för cyklister kan dela vissa likheter med de som lockar fotgängare:du vet, glassställ, trafikljus, sånt."
Modellens skapare har visat att BiteNetPp konsekvent överträffar befintliga toppmoderna metoder genom att jämföra deras förutsägelser för de protein-peptidbindningsställen som är kända genom experimentella observationer. Viktigt, den nya modellen tar mindre än en sekund att analysera en enda proteinstruktur, vilket gör den väl lämpad för storskaliga studier. Det finns tusentals protein-proteininteraktioner som potentiellt kan riktas mot peptidbaserade läkemedel, så beräkningsmetoder måste vara tillräckligt snabba för att göra deras screening genomförbar i ett farmakologiskt sammanhang.