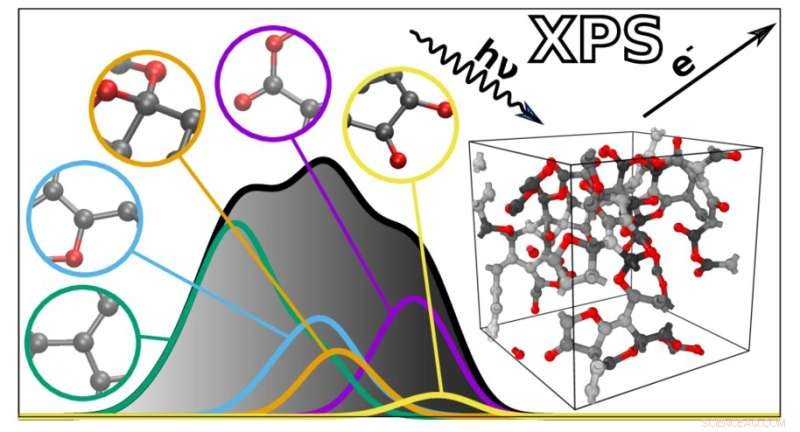
Den nya algoritmen förutsäger XPS-spektra för komplexa material baserat på individuella atombidrag. Kredit:Miguel Caro / Aalto University
Kolbaserade material har en enorm potential för att bygga en hållbar framtid, men materialforskare behöver verktyg för att korrekt analysera deras atomstruktur, vilket bestämmer deras funktionella egenskaper. Röntgenfotoelektronspektroskopi (XPS) är ett av verktygen som används för att göra detta, men XPS-resultat kan vara utmanande att tolka. Nu har forskare vid Aalto utvecklat ett maskininlärningsverktyg för att förbättra XPS-analyser, som de har gjort fritt tillgängligt som XPS Prediction Server.
XPS-spektra är grafer med en samling toppar som reflekterar bindningsenergin hos elektronerna djupt inne i atomerna som utgör ett material. Eftersom bindningsenergierna beror på atommiljön kan de användas för att sluta sig till hur atomerna är anslutna i ett visst material eller molekyl. Detta gör dock också XPS-spektra svårtolkade, eftersom många faktorer påverkar bindningsenergierna. Bindningsenergierna för olika atomära egenskaper kan också överlappa varandra, vilket ytterligare komplicerar analysen.
För att hjälpa till med detta utvecklade ett team under ledning av Miguel Caro en beräkningsmetod som kan förutsäga bindningsenergispektrumet för ett material baserat på en datorgenererad strukturmodell. Detta förenklar XPS-datatolkningen genom att göra det möjligt att matcha de experimentellt observerade bindningsenergierna mot beräkningsförutsägelserna.
Idén i sig är inte ny, men problemet har varit beräkningssvårigheten att exakt beräkna XPS-spektrumet för ett material. Caros team löste detta med hjälp av maskininlärning. Tricket var att träna en billig datoralgoritm för att förutsäga resultatet av en beräkningsdyr referensmetod baserad på en effektiv kombination av beräkningsmässigt billiga och dyra kvantmekaniska data.
Den beräkningsmässigt billigare metoden, DFT, matchar inte experimentella resultat särskilt exakt. Den mer exakta metoden, GW, tar för lång tid att beräkna när en molekyl har många atomer. "Vi bestämde oss för att konstruera en baslinjemodell som använder rikligt med DFT-data och sedan förfina den med knappa och värdefulla GW-data. Och det fungerade", säger Caro.
Den resulterande algoritmen kan förutsäga spektrumet av alla oordnade material gjorda av kol, väte och syre. "De förutsagda spektra ligger anmärkningsvärt nära de som erhålls experimentellt. Detta öppnar dörren till bättre integration mellan experimentell och beräkningsmässig karakterisering av material", säger Caro. Därefter planerar teamet att utöka sin teknik till att omfatta ett bredare utbud av material och andra typer av spektroskopi.
Artikeln med öppen tillgång publicerades i Chemistry of Materials . + Utforska vidare