Att låsa upp biologisk information från komplexa encelliga genomiska data har precis blivit enklare och mer exakt, tack vare det innovativa scLENS-verktyget som utvecklats av Biomedical Mathematics Group inom IBS Center for Mathematical and Computational Sciences som leds av chefsutredaren Kim Jae Kyoung, som också är professor vid KAIST. Detta representerar ett betydande steg framåt inom området encellig transkriptomik.
Forskningen är publicerad i tidskriften Nature Communications .
Encellig genomisk analys är en avancerad teknik som mäter genuttryck på individuell cellnivå och avslöjar cellulära förändringar och interaktioner som inte är observerbara med traditionella genomiska analysmetoder. När den tillämpas på cancervävnader kan denna analys avgränsa sammansättningen av olika celltyper i en tumör, ge insikter i hur cancer fortskrider och identifiera nyckelgener som är involverade under varje stadium av progressionen.
Trots den enorma potentialen hos encellig genomisk analys har det alltid varit utmanande att hantera den stora mängden data som den genererar. Mängden data täcker uttrycket av tiotusentals gener över hundratals till tusentals enskilda celler. Detta resulterar inte bara i stora datamängder utan introducerar också brusrelaterade distorsioner, som delvis uppstår på grund av nuvarande mätningsbegränsningar.
Motsvarande författare Kim Jae Kyoung lyfte fram:"Det har skett ett anmärkningsvärt framsteg inom experimentell teknik för att analysera encelliga transkriptomer under det senaste decenniet. Men på grund av begränsningar i dataanalysmetoder har det förekommit en kamp för att fullt ut utnyttja värdefull data som erhållits genom omfattande kostnader och tid."
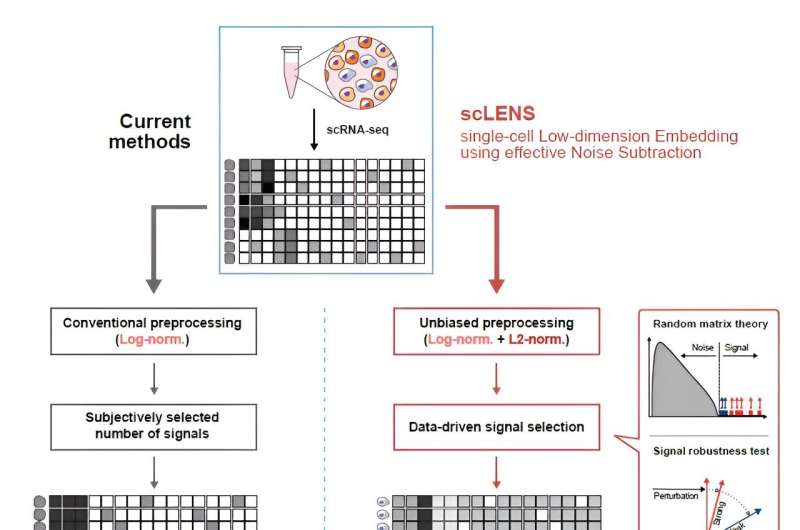
Forskare har utvecklat många analysmetoder under åren för att urskilja biologiska signaler från detta brus. Noggrannheten hos dessa metoder har dock varit mindre än tillfredsställande. En kritisk fråga är att bestämning av signal- och bruströsklar ofta beror på subjektiva beslut från användarna.
Det nyutvecklade scLENS-verktyget utnyttjar Random Matrix Theory och Signal robusthetstester för att automatiskt skilja signaler från brus utan att förlita sig på subjektiv användarinput.
Första författaren Kim Hyun konstaterade, "Tidigare var användarna tvungna att godtyckligt bestämma tröskeln för signal och brus, vilket äventyrade reproducerbarheten av analysresultat och introducerade subjektivitet. scLENS eliminerar detta problem genom att automatiskt detektera signaler med hjälp av endast den inneboende strukturen av data."
Under utvecklingen av scLENS identifierade forskare de grundläggande orsakerna till felaktigheter i befintliga analysmetoder. De fann att vanliga dataförbehandlingsmetoder förvränger både biologiska signaler och brus. Den nya förbearbetningsmetod som scLENS erbjuder är fri från sådana förvrängningar.
Genom att lösa problem relaterade till bruströskel som bestäms av subjektiva användarval och signalförvrängning i konventionell dataförbearbetning, överträffar scLENS avsevärt befintliga metoder i noggrannhet. Dessutom automatiserar scLENS den mödosamma processen för val av signaldimension, vilket gör att forskare kan extrahera biologiska signaler bekvämt och automatiskt.
Ci Kim tillade, "scLENS löser stora problem inom encellig transkriptomdataanalys, vilket avsevärt förbättrar noggrannheten och effektiviteten genom hela analysprocessen. Detta är ett utmärkt exempel på hur grundläggande matematiska teorier kan driva innovation inom biovetenskaplig forskning, vilket gör det möjligt för forskare att mer snabbt och exakt svara på biologiska frågor och avslöja livets hemligheter som tidigare var gömda."
Mer information: Hyun Kim et al, scLENS:datadriven signaldetektion för opartisk scRNA-seq dataanalys, Nature Communications (2024). DOI:10.1038/s41467-024-47884-3
Journalinformation: Nature Communications
Tillhandahålls av Institute for Basic Science