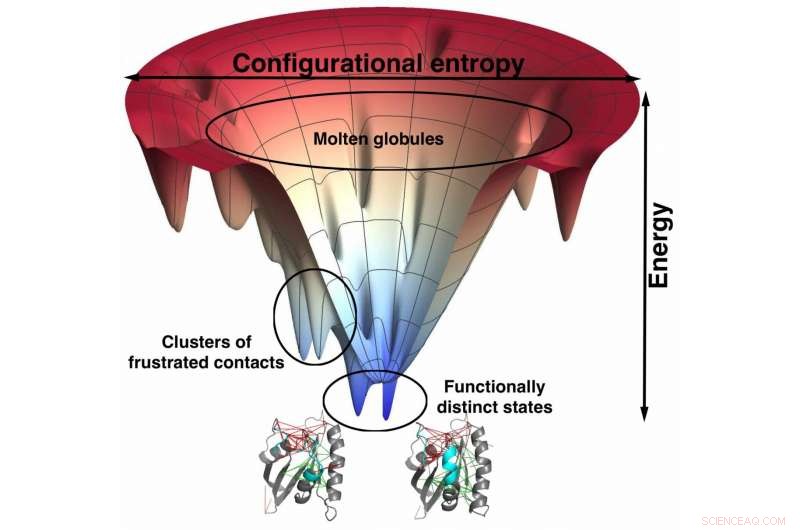
Atom-skala modeller av Rice University forskare baserade på de som används för att förutsäga hur proteiner veck visar en stark korrelation mellan minimalt frustrerade bindningsställen och läkemedelsspecificitet. Tratten, en visuell representation av proteinets energilandskap när det veck, hjälper till att hitta de frustrerade webbplatserna. Sådana modeller kan leda till bättre utformade läkemedel med färre biverkningar. Kredit:Mingchen Chen/Rice University
Att veta exakt var proteiner är frustrerade kan gå långt mot att göra bättre läkemedel.
Det är ett resultat av en ny studie av forskare från Rice University som letar efter mekanismerna som stabiliserar eller destabiliserar viktiga delar av biomolekyler.
Atomskalamodeller av risteoretikern Peter Wolynes, huvudförfattare och alumn Mingchen Chen och deras kollegor vid Center for Theoretical Biological Physics visar att inte bara vissa specifika frustrerade sekvenser i proteiner är nödvändiga för att de ska fungera, att hitta dem ger också ledtrådar för att uppnå bättre specificitet för läkemedel.
Den kunskapen kan också hjälpa till att designa läkemedel med färre biverkningar, Sa Wolynes.
Teamets studie med öppen tillgång visas i Naturkommunikation .
Modellerna i atomskala tar inte hänsyn till interaktionerna inom möjliga bindningsställen snarare än den stora majoriteten av interaktionerna i proteiner som styr deras veckning. De finare upplösningsmodellerna möjliggör införlivande av samfaktorer som kemiskt aktiva ligander, inklusive läkemedelsmolekyler. Forskarna säger att denna förmåga ger ny inblick i varför ligander bäst fångas endast av specifika proteiner och inte av andra.
"Onaturliga ligander, "aka droger, tenderar att binda bäst med de frustrerade fickorna i proteiner som blir minimalt frustrerade när läkemedlen binder, Sa Wolynes. Att ha ett sätt att hitta och sedan lära sig detaljerna om dessa minimalt frustrerade webbplatser skulle hjälpa läkemedelsföretagen att eliminera en hel del trial and error.
"Standardsättet att göra läkemedelsdesign är att prova 10, 000 bindningsställen på ett protein för att hitta de som passar, " sa Wolynes. "Vi säger att du inte behöver prova alla möjliga bindningsställen, bara ett rimligt antal för att förstå statistiken över vad som kan fungera i lokala miljöer.
"Det är skillnaden mellan att göra en omröstning och att faktiskt ha ett val, " sade han. "Undersökningen är billigare, men du måste fortfarande kolla upp saker."
Risforskarna är kända för sin energilandskapsteori om hur proteiner viker sig. Den använder vanligtvis grovkorniga modeller där aminosyror representeras av bara ett fåtal platser.
Den strategin tar mindre datorkraft än att försöka bestämma positionerna över tiden för varje atom i varje rest, och ändå har det visat sig mycket exakt när det gäller att förutsäga hur proteiner viker sig baserat på deras sekvenser. Men för denna studie, forskarna modellerade proteiner och protein-ligandkomplex på atomnivå för att se om de kunde hitta hur frustration ger vissa delar av ett protein den flexibilitet som krävs för att binda till andra molekyler.
"En av de stora sakerna med modellering vid all atomupplösning är att den tillåter oss att utvärdera om läkemedelsmolekyler passar bra in i bindningsställen eller inte, ", sade Wolynes. "Denna metod kan snabbt visa huruvida en bindningsplats för ett visst läkemedel kommer att vara minimalt frustrerad eller kommer att förbli en frustrerad region. Om platsen fortfarande är frustrerad efter att molekylen binds, proteinet kan ordna om eller så kan läkemedlet ändra dess orientering på ett sådant sätt att det kan ge upphov till biverkningar. "
Genom att modellera de frustrerade platserna - och ibland ändra dem för att se vad som skulle hända - låter forskarna se hur läkemedelsspecificitet korrelerar med bindningsfickor. Frustrationsanalys, de skrev, tillhandahåller "en väg för screening för mer specifika föreningar för läkemedelsupptäckt."
"Det här konceptet med frustration fanns där i början av vårt arbete med proteinveckning, " sa Wolynes. "När vi applicerade det på riktiga proteinmolekyler, vi hittade några exempel där vikningsmekanismen bröt mot vad vi skulle förutsäga från en perfekt tratt. Sedan upptäckte vi att dessa avvikelser från trattbilden inträffade där proteinet var, faktiskt, lite frustrerad.
"Det var som undantaget som bevisar regeln, " sa han. "Något som är sant hela tiden kan vara trivialt. Men om det inte är sant 1% av tiden, det är ett problem som ska lösas, och vi har kunnat göra det med AWSEM, vår programvara för strukturförutsägelse."
Det är möjligt att utöka programvaran för att analysera frustration på atomnivå, som beskrevs av gruppen i en annan ny artikel. Men beräkningskostnaden för att spåra varje atom i ett protein är så hög att forskarna behövde ett sätt att prova rörelserna i specifika regioner där frustration kan förvirra veckningsvägen.
"Mingchen insåg att det fanns en effektiv algoritm för att prova de lokala miljöerna på bindningsställen men behålla den atomistiska upplösningen, sa Wolynes, som noterade han och Chen, nu i privat industri, använder modellerna för att undersöka möjliga behandlingsmetoder, inklusive covid-19-relaterade läkemedel.