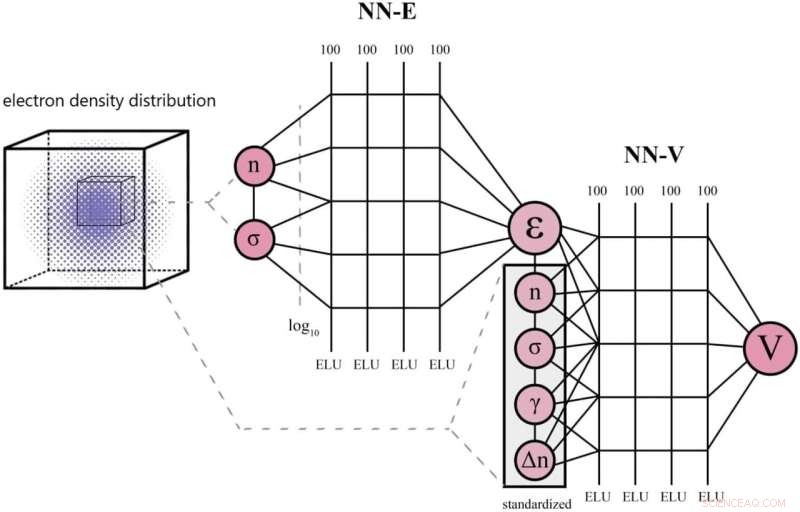
Topologi av XC neurala nätverk. Den består av två delar:NN-E förutsäger εxc och NN-V förutsäger vxc . Varje del av det neurala nätverket består av 4 lager vardera med 100 neuroner. För båda delarna behövs information om lokal densitet och dess derivator. Kredit:Scientific Reports (2022). DOI:10.1038/s41598-022-18083-1
Forskare från Center for Materials Technologies vid Skoltech har levererat en proof-of-concept-demonstration av en neural nätverksdriven metod för att skapa en exakt utbytes-korrelationsfunktionell interpolation, som är kärnkomponenten i densitetsfunktionella teorin. DFT är i sin tur den huvudsakliga numeriska metoden som används inom kondenserad materiens fysik och kvantkemi för att beräkna föreningsreaktivitet, molekylers zonstruktur, material hållbarhet och andra egenskaper som är avgörande för sökandet efter nya material, läkemedel och mer. Den lovande neurala nätverksarkitekturen presenterades och analyserades i Scientific Reports .
Som beskrivs av multielektron Schrödinger-ekvationen, bestämmer elektronernas rörelser i materia egenskaperna hos den elektroniska strukturen. Till exempel är den kemiska bindningen, ett kärnbegrepp i all kemi, en komplex korrelerad rörelse av elektroner som styrs av kvantmekanikens lagar.
Problemet med multielektron Schrödinger-ekvationen är att även om det är relativt lätt att ange, har ingen analytisk lösning hittats, och den numeriska lösningen är mycket komplex och utmanande. Här är ett av de viktigaste tillvägagångssätten medelfältsmetoden (densitet), som beskriver den komplexa interaktionen mellan elektroner i termer av en effektiv potential.
"Densitetsfunktionella teorin förenklar saker genom att använda föreställningen om ett elektronmoln som kännetecknas av viss lokal täthet istället för att överväga individuella elektroner," förklarade den första författaren till studien, Skoltech Research Engineer Alexander Ryabov.
"Denna teori har dock ett viktigt okänt värde, kallat utbytes-korrelationsfunktionen. Fram till nyligen var tendensen att approximera den analytiskt. Det vill säga att koefficienter i den funktionella formen bestämdes utifrån flera kända fysiska principer utan att använda neurala nätverk . Vår metod är den första som använder ett tvåkomponents neuralt nätverk för detta. Neurala nätverk har använts aktivt i denna uppgift, men vårt team banar väg för dem på detta område i Ryssland."
Enligt forskarna är det som skiljer deras från de konkurrerande metoderna att träningen sker i två steg:Först tränas ett nätverk och dess vikter fryses. Sedan lärs en till.
"Tidigare använde människor ett neuralt nätverk för att approximera utbytes-korrelationsfunktionen, varefter beräkningsintensiva derivator måste tas för att hitta motsvarande utbyteskorrelationspotential. Det här är derivator av ett slag som ofta visar sig vara svåra att beräkna med hyfsad noggrannhet med hjälp av ett neuralt nätverk", tillade Skoltech Senior Research Scientist Petr Zhilyaev, studiens huvudutredare. "I vårt arbete approximerar ett tvåkomponents neuralt nätverk både potentialen och den funktionella, så inga komplicerade derivator är inblandade, och beräkningsbelastningen minskar."
"För att köra experimenten som rapporterats i vår artikel, implementerade vi det neurala nätverket i Octopus mjukvarupaket för kvantkemi," sa Ryabov. "Vi undersökte också hur träningsprocessen påverkas av icke-självkonsistenta tätheter. Efter att ha lagt till sådana tätheter i träningsdatasetet, observerade vi förbättrad prestanda på molekyler för vilka det neurala nätverket tidigare gav de sämsta resultaten." + Utforska vidare