Grafiskt abstrakt. Kredit:Journal of Molecular Biology (2022). DOI:10.1016/j.jmb.2022.167525
Vilka gener är specifika för en viss celltyp, det vill säga "markerar" deras identitet? Med den ökande storleken på datauppsättningar nuförtiden är det ofta svårt att svara på denna fråga. Ofta är markörgener helt enkelt gener som har hittats i specifika cellpopulationer. Men många fler gener kan vara karakteristiska för en viss celltyp men förbli oupptäckta.
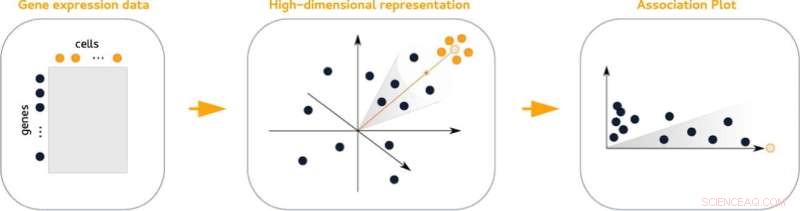
Association plots (APL), en ny statistisk metod för att visualisera genaktivitet inom ett cellkluster, gör det lättare att hitta dess markörgener. Diagrammen jämför aktiviteten hos gener i ett givet kluster med alla andra kluster från datamängden. Dessutom gör de det enkelt att se vilka gener som delas med andra kluster.
"Associationsdiagram tillåter oss inte bara att identifiera nya markörgener. Det fungerar också tvärtom - vi kan matcha kluster av okänd identitet i en datauppsättning med celltyper, baserat på en tillhandahållen lista över markörgener", säger Elzbieta Gralinska vid Max Planck Institute for Molecular Genetics i Berlin.
Bioteknologen arbetar i teamet med Martin Vingron, som utvecklade tekniken. Forskarna visade teknikens funktionalitet på två allmänt tillgängliga datauppsättningar och publicerade resultaten i Journal of Molecular Biology . Dessutom har APL släppts som en gratis modul för den statistiska miljön R. APL-paketet gör det möjligt för forskare att visuellt inspektera sina encellsdata och välja individuella gener med markören för att lära sig mer djupgående detaljer.
Analysera och gruppera enskilda celler
Varför är det nödvändigt att identifiera markörgener i första hand? Moderna sekvenseringsteknologier kan dechiffrera individuella RNA-molekyler i enskilda celler. Från ett blodprov kan till exempel varje cell separeras och ett prov av cellens RNA kan avkodas. Dessa encellsdata representerar de aktiva generna som transkriberades till RNA-molekyler.
Fördelen:Istället för att fundera över vilken celltyp ett visst RNA tillhör, kan det spåras tillbaka till sin ursprungscell. Nackdelen:att sekvensera tusentals RNA i varje enskild cell av tiotusentals celler producerar extraordinära mängder data.
En utväg är att sortera cellerna utifrån deras RNA-innehåll. "Encellsdata är sammansatta av en vild blandning av många olika celltyper. Vi är intresserade av celler av samma celltyp, som alla borde bete sig likadant", förklarar Martin Vingron. Därför är det vettigt att gruppera liknande celler beräkningsmässigt, säger han. "För oss definierar markörgenerna en celltyp."
Utforska cellkluster interaktivt
Med hjälp av allmänt tillgängliga data från vita blodkroppar visade teamet hur den nya algoritmen fungerar. De många olika typerna av vita blodkroppar som T-celler, B-celler eller monocyter är alla grupperade i separata kluster. Forskarna bekräftade kända markörgener och kunde visa att nära släktingar bland blodkropparna också delar stor likhet i sin genaktivitet.
"Var och en av markörgenerna vi hittade med APL kunde ha upptäckts av åtminstone en annan befintlig metod för identifiering av markörgener," säger Gralinska. Men fördelen med APL framför de befintliga algoritmerna är dess grafiska representation av resultaten, säger hon. "Befintliga verktyg ger långa listor med gener och poängvärden. Ofta går användare igenom listan och stannar vid en godtycklig gräns."
Däremot ger den nya metoden ett sätt att visualisera dessa gener, klicka på var och en och titta närmare på dess aktivitet, säger hon. "Vi tillhandahåller inte bara listor över markörgener, vi tillåter användare att granska hur dessa gener beter sig", säger forskaren. "Med associationsdiagram kan de dyka in i sina data för att lära sig mer om varje celltyp." Dessutom, säger hon, är det väldigt lätt att bryta ner den biologiska rollen för de mest intressanta generna i ett efterföljande steg via Gene Ontology term anrikningsanalys, som är kompatibel med APL-mjukvaran – något hon anser vara "en mycket användbar funktion."
Den underliggande matematiska modellen
De högdimensionella data som innehåller information om aktivitet över gener kan inte representeras visuellt utan förlust av information. Detsamma gäller för klustrade data, som allt komplicerar analysen. "Vårt knep är att vi tar hänsyn till många fler än bara två eller tre dimensioner, men i slutändan skapar ett tvådimensionellt diagram", säger Gralinska.
Associationsplotterna härrör från en matematisk teknik som samtidigt bäddar in både gener och celler i ett gemensamt, högdimensionellt utrymme. Att mäta avstånden mellan gener och ett givet cellkluster i detta utrymme resulterar i värdepar som återspeglar associationen av en gen till ett givet kluster och ger insikter om dess association till andra kluster.
"En brist med APL är att vi förlitar oss på för-klustrade data, vilket innebär att vi måste förlita oss på andra tekniker för klustring", säger Martin Vingron. "Ändå hoppas vi att vår nya metod kommer att hitta många nya användare. Vi tycker att en visuell och interaktiv process helt enkelt gör en bättre analys."