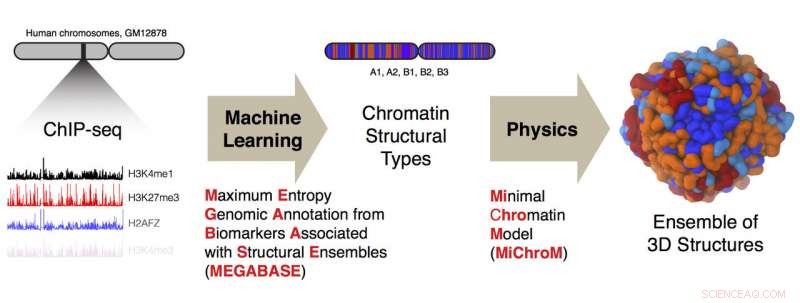
Forskare vid Rice University och Baylor College of Medicine har utvecklat en beräkningspipeline för att omvandla endimensionell ChIP-sekvenseringsdata om DNA till tredimensionella strukturer av mänskliga kromosomer. Kredit:Ryan Cheng/Michele Di Pierro
DNA:t i en mänsklig cell är 2 yards (1,83 meter) långt och sveper runt miljontals pärlliknande histonproteiner för att passa in i cellens kärna. Forskare vid Rice University och Baylor College of Medicine visade att att undersöka det kemiska tillståndet hos dessa proteiner gör det möjligt att förutsäga hur en hel DNA-kromosom kommer att vikas.
Forskare baserade vid Rice's Center for Theoretical Biological Physics (CTBP) har konstruerat datormodeller för att analysera epigenetiska märken, som inkluderar proteiner bundna till DNA såväl som kemiska modifieringar av histonproteiner. De skördade informationen som kodades i dessa markeringar för att förutsäga hur kromosomerna viker sig i tre dimensioner.
Deras fynd flyttar genetikområdet närmare förmågan att förutsäga den vikta strukturen av hela genom, som en dag skulle kunna hjälpa till att identifiera felveckningsrelaterade genetiska sjukdomar.
Verket visas denna vecka i Förfaranden från National Academy of Sciences .
Packad in i kärnan, DNA viks till en funktionell form som skiljer sig åt i olika typer av celler. Eftersom varje cell i en organism innehåller samma DNA, epigenetiska märken hjälper den att hitta rätt form för den typ av cell den bor i.
"Något ovanpå den genetiska koden talar om för cellen vad den ska vara och bestämmer vilka delar av kromosomen som ska läsas vid varje given tidpunkt, " sa biofysikern Peter Wolynes, en medförfattare till tidningen. "Det här är de så kallade epigenetiska märkena."
Kollektivt, epigenetiska märken hjälper till att packa in genomet i de lösa men välorganiserade fack som det antar under interfas, den arbetande "medelåldern" i en cells liv. Dessa fack bringar transkriptionsrelaterade gener nära varandra och tillåter dem att kommunicera och fungera.
Epigenetiska märken kan avslöjas med en etablerad teknik som kallas ChIP-sekvensering, som kartlägger proteinbindningsställen längs DNA.
"Vi förstår inte exakt hur genomet markeras, men vi kan mäta det genom chip-sekvensering, som har blivit ett ganska okomplicerat experiment, " Sa Wolynes. "På samma sätt som vi kan se genetisk kod (DNA), vi kan också mäta dessa märken direkt i många olika celler. De har blivit nästa sekvenslager i genomet."
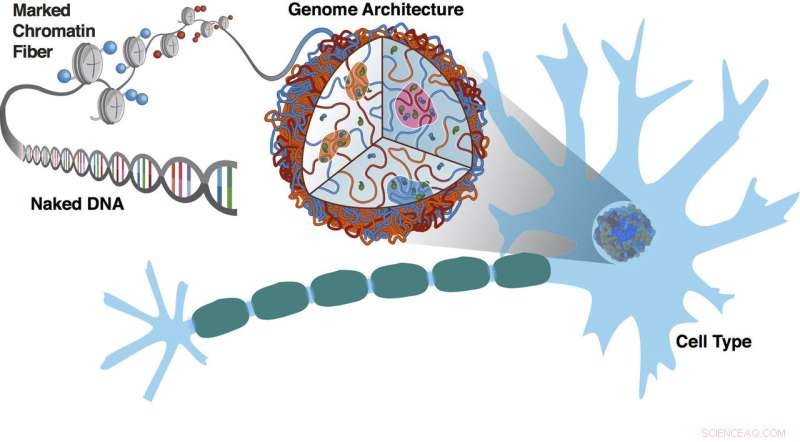
Experiment vid Rice University och Baylor College of Medicine visar hur segment av kromatin med samma epigenetiska markeringsmönster lokaliseras tillsammans i en process relaterad till fasseparation. Naket DNA är dekorerat av epigenetiska markeringar som kodar för det tredimensionella arrangemanget av kromosomer. Genomarkitekturen och markeringsmönstren är egenskaper hos celltypen, i detta fall en nervcell med dess karakteristiska myelinskida. Kredit:Sigrid Knemeyer/Centrum för teoretisk biologisk fysik vid Rice University
"Det är en annan nivå av information, " sa medförfattaren och biofysikern José Onuchic. "Alla dina cellers DNA är detsamma. Dock, olika sorters celler har olika epigenetik, så deras uttrycksmönster är olika."
Co-lead författare och Rice postdoktorala stipendiater Michele Di Pierro och Ryan Cheng använde ChIP-sekvenseringsdata för en human lymfoblastcell som sonderar 84 olika DNA-bindande proteiner och 11 kemiska modifieringar av histoner. Histonproteiner hjälper till att organisera genomet genom att fungera som spolar runt vilka DNA lindas.
Med hjälp av data från bara några av kromosomerna, de tränade ett anpassat neuralt nätverk som heter MEGABASE (Maximum Entropy Genomic Annotation from Biomarkers Associated with Structural Ensembles) för att producera en sekvens av kromatintyper. Det avslöjade hur de epigenetiska märkena var relaterade till avdelningarna, sa de. Väl utbildad, de validerade MEGABASE-modellen genom att mata den med data från de återstående kromosomerna. Det gav en ny uppsättning strukturtyper för analys av Rice-teamets MiChroM-program, en kusin till labbets AWSEM energilandskapsalgoritm som förutsäger strukturerna hos proteiner. MiChroM-algoritmen förutspådde 3D-strukturerna hos kromosomerna.
"Våra fynd stöder tanken att kompartmentalisering i kromosomer uppstår från fasseparationen av olika kromatintyper i kärnan, som separeringen av olja och vatten, " sa Cheng.
När forskarna reducerade den ursprungliga datamängden till bara de 11 histonmarkeringarna och körde beräkningarna igen, resultaten var bara marginellt annorlunda. I sista hand, de fastställde att enbart histondata är tillräckliga för att förutsäga en kromosoms form. "Det finns en väldefinierad kod som kopplar histonmarkeringarna till strukturen, " sa Di Pierro. "Det är välbevarat, så det är troligt att det har en funktion."
För att validera deras teori, forskarna jämförde sina resultat med kontaktkartor över lymfoblastceller genererade av Hi-C. Denna experimentella teknik, som använder sekvensering med hög genomströmning för att identifiera vikningsmönster i DNA, utvecklades av medförfattaren Erez Lieberman Aiden, chef för Baylor's Center for Genome Architecture och senior utredare vid CTBP.
"Det här dokumentet säger att vi kan ta endimensionell information om histoner och använda den med våra big-data-verktyg för att förutsäga tredimensionell struktur, " sa Wolynes.
Deras framgång för teamet närmare det slutliga målet för en teori som förutsäger arkitekturen för ett helt genom. Dock, ett kyckling-eller-ägg-problem kvarstår:Vikas kromatinet på grund av markörerna, eller visas markörerna på grund av vikningen?
"Det är en del av vår fascination av hur livet fungerar, " sa Di Pierro. "Det är ett vackert problem."